





















OpenAI son zamanlarda birçok kez farklı yeni modeller sundu ve bu modellerin hiçbiri kamuoyuna açıklanmadı....
Sci. Adv. | Viral Yapının Evrensel Kodu: AlphaFold Veritabanı, Üç Alanlı Virüsler Arasındaki Ortak Protein Katlanma Kalıplarını Ortaya Çıkarıyor
Yazı Özetini Göster


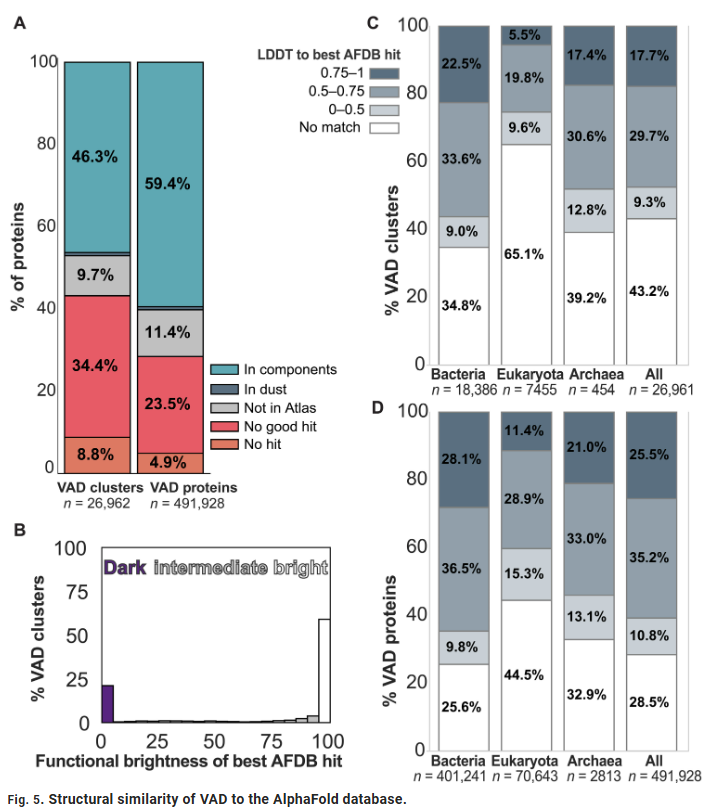
Araştırmacılar, virüs monomerlerini ve homolog dimerlerini kapsayan bir yapısal tahmin veritabanı oluşturmak için **AlphaFold**’u kullandılar ve bu sayede virosferdeki korunan protein katlanmalarının çeşitliliğini ve dağılımını sistematik olarak ortaya çıkarmayı amaçladılar. Bakteriyel, arkeal ve ökaryotik konaklardan elde edilen 200 binden fazla viral proteinin yapısal modellemesi ve kümeleme analizi sonucunda, araştırmacılar birçok katlanma yapısının farklı konak virüslerinde, hatta viral sınıflandırma sınırları boyunca bile kayda değer bir **korunum** sergilediğini keşfetti. Bu katlanmaların bir kısmı, konak proteinleriyle homolog yapılar paylaşmakta, bu da virüslerin evrim sırasında gen değişimi ve katlanma yeniden kullanımı yoluyla yapısal **”evrensel modüller”** oluşturduğunu düşündürmektedir. Bu çalışmada oluşturulan **Viral AlphaFold Veritabanı (VAF-DB)**, sistematik ilk viral yapısal tahmin kaynağı olarak, viral protein fonksiyonu çıkarımı, köken araştırmaları ve antiviral ilaç hedefi keşfi için yeni bir araç sunmaktadır.
Virüsler, yeryüzündeki en bol ve çeşitli biyolojik varlıklardır; kodladıkları proteinler ise son derece geniş bir kimyasal ve fonksiyonel alanı kapsar. Evrimsel açıdan bakıldığında, viral genomların çeşitliliği ve konaklarla olan gen alışverişleri, yaşamın erken dönem evriminde kilit rol oynamıştır. Bununla birlikte, hücresel organizmalarla kıyaslandığında, viral yapısal bilgi son derece sınırlıdır ve viral proteinlerin yaklaşık %95’inin deneysel olarak çözülmüş üç boyutlu yapısı bulunmamaktadır. Bu boşluk, araştırmacıların viral fonksiyon ve evrim mekanizmalarını anlamasını kısıtlamaktadır.
**AlphaFold**’un ortaya çıkışı, protein yapı araştırmalarının çehresini büyük ölçüde değiştirmiştir. Monomerik protein yapı tahminindeki yüksek doğruluğu, birden fazla genom veritabanında doğrulanmıştır. Araştırmacılar bu temelde, farklı virüsler arasındaki katlanma ortaklıklarını ve evrimsel ilişkileri keşfetmek için derin öğrenme yapı tahminini tüm viral proteomuna genişletmeyi önermişlerdir.
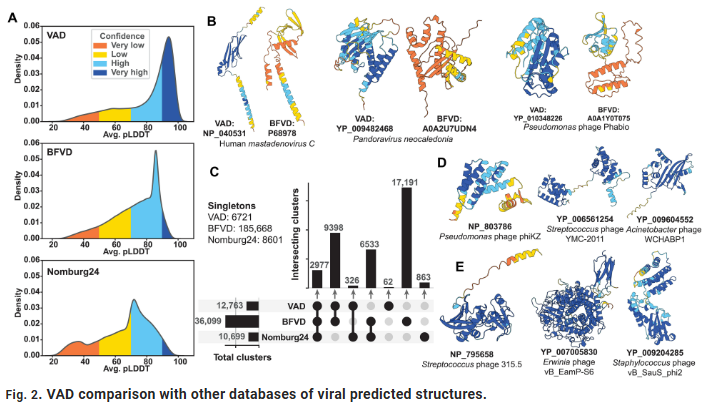
Bazı viral protein veritabanları az sayıda tahmin edilmiş yapı içerse de, özellikle bakteriyel ve arkeal virüsler olmak üzere, tüm konak kaynaklı virüsleri sistematik olarak kapsayan birleşik bir veri kaynağı henüz mevcut değildir. Bu nedenle, araştırmacılar konaklar arası viral yapısal korunumu ve potansiyel evrimsel önemini analiz etmek için monomer ve homolog dimer tahmin sonuçlarını kapsayan büyük ölçekli bir **Viral AlphaFold Veritabanı** oluşturmuşlardır.
Yöntem
Araştırmacılar, birden fazla yetkili viral genom kaynağından (NCBI RefSeq, IMG/VR ve GenBank dahil) yaklaşık 220 bin tekrarsız viral protein dizisini birleştirmiş ve bunları konak türlerine göre üç ana kategoriye ayırmıştır: **bakteriyel virüsler, arkeal virüsler ve ökaryotik virüsler**. Tüm diziler, uzunluk ve kalite filtrelemesinden geçtikten sonra, monomer ve homolog dimer tahmini için **AlphaFold2** ve **AlphaFold-Multimer** kullanılmıştır.
Tahmin edilen modeller, kalite puanına (pLDDT ve pTMscore) göre filtrelenerek kümeleme analizi için yüksek güvenilirliğe sahip yapılar korunmuştur. Ardından, araştırmacılar yapısal benzerliğe (TM-align ve Foldseek) dayalı bir viral protein yapısal ağı oluşturmuş ve kümeleme ve hizalama yoluyla konaklar arası korunan katlanma tiplerini tanımlamışlardır. Ek olarak, olası yapısal miras ve fonksiyonel homoloji ilişkilerini keşfetmek için bu katlanmalar bilinen PDB girişleri ve konak proteinleriyle karşılaştırılmıştır.
Elde edilen sonuçlar **VAF-DB** platformuna kaydedilmiş ve etkileşimli bir arayüz aracılığıyla görselleştirme, indirme ve açıklama işlevleri sunulmuştur.
Sonuçlar
Viral Protein Katlanma Alanının Genel Dağılımı
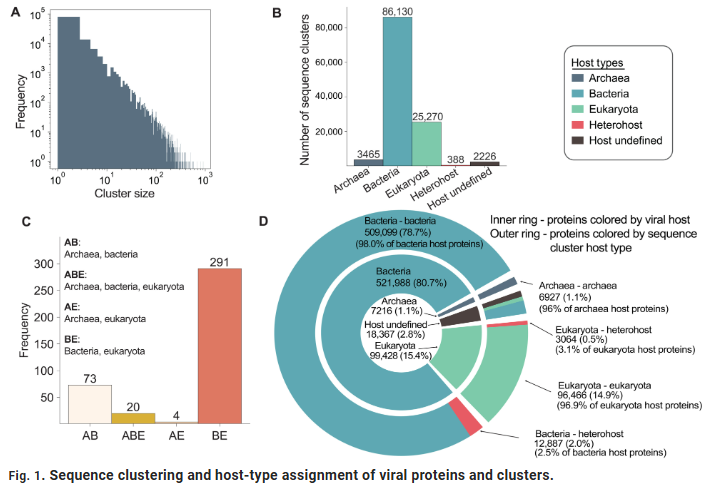
Tahmin edilen 200 binden fazla yapıdan, modellerin yaklaşık %67’si yüksek güvenilirliğe sahiptir (ortalama pLDDT > 80). Viral monomerik katlanma alanı oldukça yoğun bir dağılım göstermekte; viral proteinlerin yaklaşık %30’u 2,000’den az yapısal kümeye ayrılabilmektedir. Bu, viral genetik çeşitliliğe rağmen, yapısal düzeyde belirgin bir **yakınsama** olduğunu göstermektedir.
Bu katlanmalar başlıca nükleik asit bağlama alanlarını, sarmal demet yapılarını, $\beta$-fıçı yapılarını ve Rossmann benzeri nükleotit bağlama yapılarını içermekte olup, virüslerin evrim boyunca az sayıda yüksek kararlılığa sahip katlanma çerçevesini tekrar tekrar kullandığını düşündürmektedir.
Konaklar Arasında Korunan Katlanmaların Keşfi
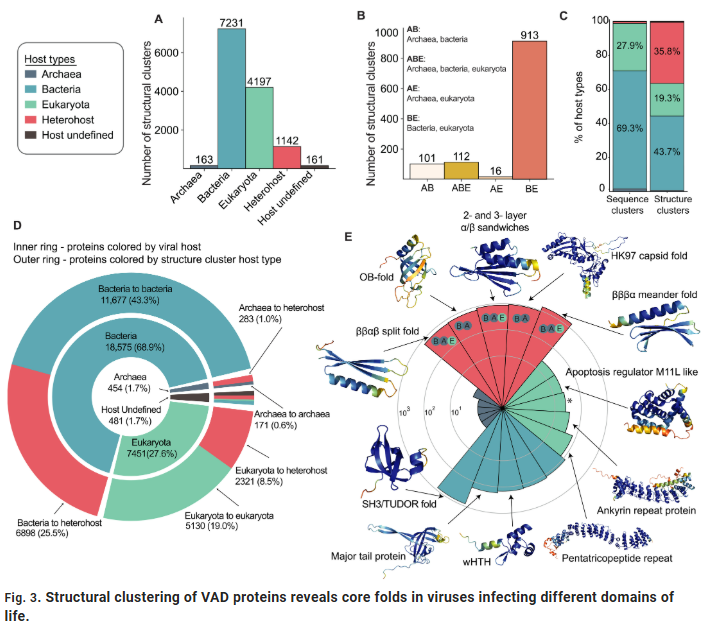
Araştırmacılar, viral yapısal kümeleme analizinde konaklar arası korunan 500’den fazla katlanma kümesi belirledi; bunlardan 72’si aynı anda bakteriyel, arkeal ve ökaryotik virüslerde bulunmaktadır. Bu katlanmalar, güçlü yapısal korunumu gösteren **ATPaz çekirdek yapılarını, metal iyonu bağlama alanlarını ve nükleik asit tanıma modüllerini** içermektedir.
Örneğin, viral kapsid proteinlerindeki **HK97 katlanması** birçok faj ve hayvan virüsünde bulunmuştur; Rossmann benzeri yapı ise nükleotit metabolizmasıyla ilgili proteinlerde yaygın olarak korunmuştur. Bu türler arası yapıların varlığı, virüslerin yaşamın erken evriminde **ortak bir katlanma “gen havuzu”** oluşturduğunu düşündürmektedir.
Virüs ve Konak Arasındaki Yapısal Homoloji
Viral katlanmaların konak proteinleriyle olan bağlantısını araştırmak için, araştırmacılar viral katlanmaları **UniProt**’taki konak yapılarıyla karşılaştırdılar. Sonuçlar, viral katlanmaların yaklaşık %18’inin konak proteinleriyle belirgin yapısal benzerlik gösterdiğini ortaya koydu.
Tipik örneklerden biri, hem fajlarda hem de ökaryotik virüslerde benzer topolojiye sahip çinko parmak yapısı içeren DNA bağlayıcı proteinlerdir. Bu tür yapılar, yatay gen transferi veya ortak bir atadan korunma yoluyla yayılmış olabilir, bu da virüslerin evrimsel olarak konaklarla karmaşık katlanma paylaşım mekanizmalarına sahip olduğunu göstermektedir.
Homolog Dimer Yapıları Viral Proteinlerin Fonksiyonel Özelliklerini Ortaya Koyuyor
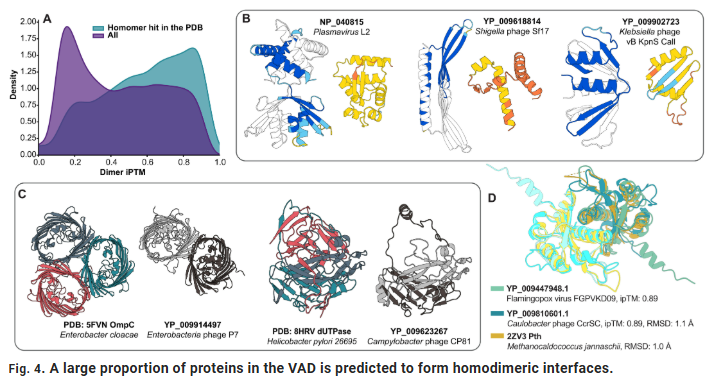
**AlphaFold-Multimer** modellemesi aracılığıyla, araştırmacılar 45 bin yüksek güvenilirliğe sahip viral homolog dimer modeli elde etti. Dimerlerin yaklaşık %60’ı, esas olarak kapsid montajı, ATP güdümlü aparatlar ve replikasyon komplekslerinde yoğunlaşan belirgin simetri (C2 simetrisi) göstermiştir.
Bu sonuçlar, virüslerin sınırlı katlanma kombinasyonları aracılığıyla karmaşık fonksiyonel kompleksler oluşturduğu varsayımını desteklemekte ve viral proteinlerin kendiliğinden birleşme ve çoklama mekanizmalarını anlamak için yapısal bir temel sağlamaktadır.
Viral Katlanmaların Fonksiyonel Açıklamaları ve Protein Ailesi Eşleştirmesi
Araştırmacılar, potansiyel fonksiyonel kategorileri belirlemek için tüm yüksek güvenilirliğe sahip katlanmaları **Pfam** ve **EC** sınıflandırma sistemleriyle eşleştirdi. Sonuçlar, viral katlanmaların başlıca beş fonksiyonel kategoriye odaklandığını gösterdi:
-
Nükleik asit bağlama ve modifikasyonu (örneğin nükleik asit helikaz, integraz);
-
Enerji metabolizması (ATPaz, dehidrogenazlar);
-
Yapı ve montaj (kapsid, iskele proteinleri);
-
Sinyalizasyon ve düzenleme;
-
Açıklanmamış veya potansiyel olarak yeni fonksiyonel katlanmalar.
Katlanmaların yaklaşık %20’si bilinen hiçbir ailede yer almamıştır, bu da virosferde büyük miktarda **”yapısal karanlık madde”**nin bulunduğunu ve işlevlerinin henüz doğrulanmadığını göstermektedir.
Viral AlphaFold Veritabanı (VAF-DB) Yapısal Görselleştirme ve Erişim Arayüzü
Araştırmacılar, yapısal görselleştirme, karşılaştırma ve toplu indirme işlevlerini sağlayan **VAF-DB** çevrimiçi platformunu (https://viral.afdb.org) geliştirmiştir.
Platform şunları destekler:
-
Konak, viral aile, fonksiyonel anahtar kelime veya yapısal benzerliğe göre arama yapma;
-
Monomer veya dimer modellerini 3 boyutlu görselleştirme;
-
Tahmin dosyalarını, kalite metriklerini ve açıklama bilgilerini indirme;
-
Farklı virüsler arasındaki yapısal benzerlik haritalarını çapraz karşılaştırma.
Platform, etkileşimli bir mimari kullanarak web arayüzünde korunan katlanma ağlarını dinamik olarak görüntüleyebilir ve bunlar arasındaki bağlantıları gösterebilir; bu da onu şu anda en kapsamlı viral yapısal tahmin kaynağı yapmaktadır.
Viral Katlanmaların Evrimsel İlişkisi ve Alanlar Arası Korunum Analizi
Yapısal benzerliğe dayalı filogenetik ağ analizi aracılığıyla, araştırmacılar bazı viral katlanmaların bakteriyel, arkeal ve ökaryotik virüsler arasında **alanlar arası bağlantı düğümleri** oluşturduğunu bulmuştur. Bu “katlanma köprüleri” esas olarak nükleik asit işleme ve montajı ile ilgili fonksiyonlarda yoğunlaşmakta, bu da virüslerin farklı konak ekolojilerinde benzer yapısal çerçeveleri tekrar tekrar kullandığını yansıtmaktadır.
Ayrıca, evrimsel ağ, bazı katlanmaların virüsler ve konaklar arasında kapalı döngü ilişkileri gösterdiğini, bunun da birden fazla **yatay gen transferi** olayının gerçekleştiğini düşündürdüğünü ortaya koymuştur. Araştırmacılar, bu türler arası korunan katlanmaların, yaşamın erken kökeninin ortak yapısal prototiplerini temsil edebileceğini ve virüsler ile hücresel yaşam arasındaki evrimsel ilişkileri yeniden inşa etmek için ipuçları sunduğunu öne sürmektedir.
Tartışma
Araştırmacılar, **Viral AlphaFold Veritabanı**nı oluşturarak virüslerin konaklar arası evrimdeki yapısal korunum ve modüler özelliklerini ortaya çıkardılar. Sonuçlar, viral protein katlanma alanının, dizilim çeşitliliğinin ima ettiğinden çok daha küçük olduğunu göstermektedir; çok sayıda farklı virüs, işlevsel çeşitliliği gerçekleştirmek için sınırlı sayıda yapısal modülü ortaklaşa kullanmaktadır.
Bu modüler katlanmalar, hem klasik kapsid ve enzimatik aktif çekirdekleri hem de daha önce açıklanmamış yeni topolojileri içermektedir. Viral katlanmaların yeniden kullanımı ve yakınsaması, virüslerin evrimsel olarak yapısal kararlılık ve montaj verimliliğinin çifte kısıtlamasına maruz kaldığını ve aynı zamanda katlanma “yeniden kullanımı” yoluyla yüksek adaptasyon ve fonksiyonel yenilik elde ettiğini göstermektedir.
**VAF-DB**’nin kurulması, viral yapısal genomik sınırlarını genişletmekle kalmamış, aynı zamanda gelecekteki fonksiyonel açıklama, protein mühendisliği ve antiviral ilaç keşfi için temel bir platform sağlamıştır. Araştırmacılar, bu veritabanının viral yapısal evrimi ve yaşamın kökenini keşfetmek için kilit bir kaynak olacağına ve yapay zeka güdümlü viral yapısal biyoloji araştırmalarını destekleyeceğine inanmaktadır.
Derleyen | DrugOne Ekibi
Referanslar
Roni Odai et al. ,The Viral AlphaFold Database of monomers and homodimers reveals conserved protein folds in viruses of bacteria, archaea, and eukaryotes.Sci. Adv.11,eadz8560(2025).
DOI:10.1126/sciadv.adz8560
İçerikte yer alan görseller telif hakkı içeriyorsa, lütfen silinmesi için bizimle iletişime geçin.
Benzer Yazılar

1.3 metre boyundaki Little Potato isimli robot, ipeksi bir pürüzsüzlükte katmanlama yapabiliyor. Yanlış anlamayın,...

Andrew Ng, yakın zamanda Y Combinator'da girişimcilik deneyimlerini paylaştığı bir konuşma yaptı. Bir girişimin başarısının...
